ATXN7 Polyclonal Antibody
For reference only. Please follow the manual included in your kit for instructions.
Catalog Number

Immunohistochemistry of paraffin-embedded Human liver cancer tissue using ATXN7 Polyclonal Antibody at dilution of 1:60(×200)
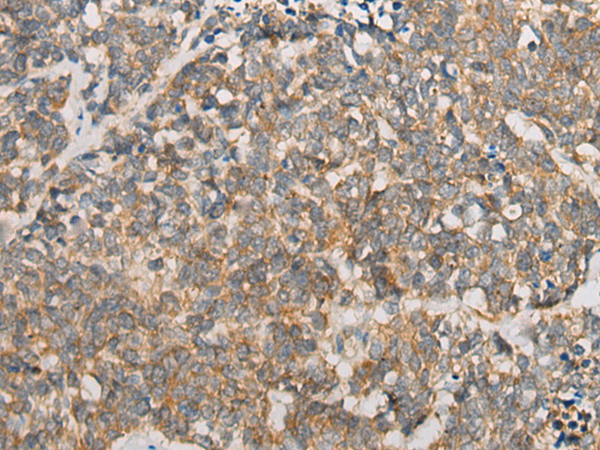
Immunohistochemistry of paraffin-embedded Human lung cancer tissue using ATXN7 Polyclonal Antibody at dilution of 1:60(×200)
Product Name
ATXN7 Polyclonal Antibody
Catalog Number
RD253584A
Clonality
Polyclonal
Purification Method
Antigen affinity purification
Isotype
IgG
Host
Rabbit
Background
The autosomal dominant cerebellar ataxias (ADCA) are a heterogeneous group of neurodegenerative disorders characterized by progressive degeneration of the cerebellum, brain stem and spinal cord. Clinically, ADCA has been divided into three groups: ADCA types I-III. ADCAI is genetically heterogeneous, with five genetic loci, designated spinocerebellar ataxia (SCA) 1, 2, 3, 4 and 6, being assigned to five different chromosomes. ADCAII, which always presents with retinal degeneration (SCA7), and ADCAIII often referred to as the 'pure' cerebellar syndrome (SCA5), are most likely homogeneous disorders. Several SCA genes have been cloned and shown to contain CAG repeats in their coding regions. ADCA is caused by the expansion of the CAG repeats, producing an elongated polyglutamine tract in the corresponding protein. The expanded repeats are variable in size and unstable, usually increasing in size when transmitted to successive generations. This locus has been mapped to chromosome 3, and it has been determined that the diseased allele associated with spinocerebellar ataxia-7 contains 38-130 CAG repeats (near the N-terminus), compared to 7-17 in the normal allele. The encoded protein is a component of the SPT3/TAF9/GCN5 acetyltransferase (STAGA) and TBP-free TAF-containing (TFTC) chromatin remodeling complexes, and it thus plays a role in transcriptional regulation. Alternative splicing results in multiple transcript variants.
Immunogen Information
Immunogen
Synthetic peptide of human ATXN7
Swissprot
O15265
Synonyms
ADCAIIATXN 7OPCA IIIOPCA3SCA 7SCA7Spinocerebellar Ataxia 7Spinocerebellar ataxia type 7 protein
Gene Accession
NP000324
Applications
Reactivity
Human, Mouse
Tested Applications
IHC,ELISA
Conjugation
Unconjugated
Dilution
IHC 1:50-1:300, ELISA 1:5000-1:10000
Concentration
0.9 mg/mL
Storage Buffer
PBS with 0.05% NaN3 and 40% Glycerol, pH7.4
Storage Instructions
Store at -20°C. Avoid freeze / thaw cycles.