
Antibody Application Techniques
May 09, 2022Reddot Biotech’s antibodies are validated for a variety of common application techniques. Always refer to the product manual that comes with your order for best results.
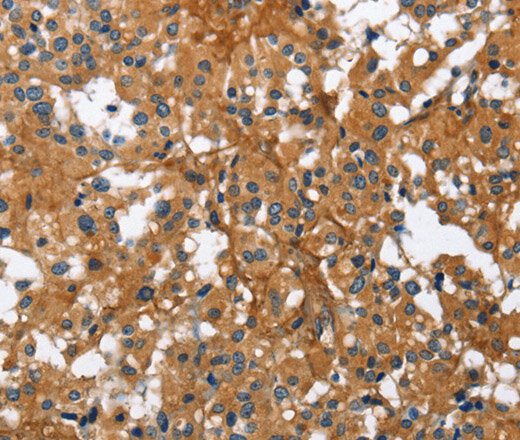
Immunohistochemistry
Immunohistochemistry (IHC) is a form of immunostaining that involves using antibodies to identify antigens in a tissue sample. Visualization of the results is either accomplished using chromogenic immunohistochemistry (light visualization) or immunofluorescence (fluorescent visualization).
The individual steps and reagents necessary for immunohistochemistry can vary widely and are dependent on the specific sample and target antigen. However, there are two main steps when preforming immunohistochemistry: sample preparation and sample labelling.
When preparing the sample, the tissue is fixed to prevent any degradation or other changes and sliced or sectioned for easier visualization.
For sample labelling, an appropriate antibody and IHC reporter must be selected. Target antigens can be detected directly or indirectly. Direct detection involves using a single step staining method where a labelled antibody is added and binds to the target antigen. Indirect detection involves using a two-step staining method where a primary antibody is added and binds to the target antigen. Then, a labelled secondary antibody binds to the primary antibody.
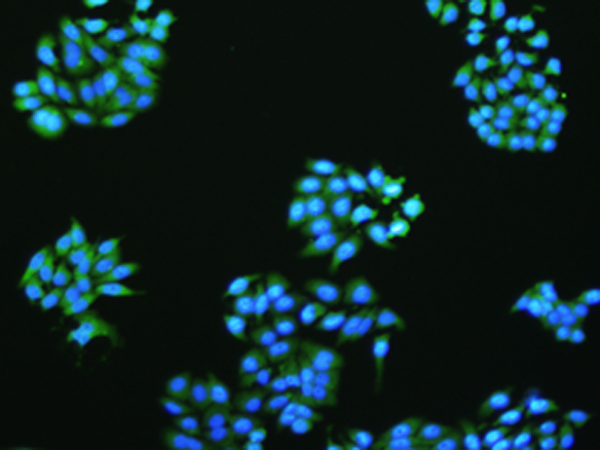
Immunofluorescence
Immunofluorescence (IF) is a technique used to visualize a target antigen bound by a fluorescent dye-labelled antibody. IF can be used on tissue samples, cultured cells lines or individual cells. The fluorophore can either be conjugated directly to the antibody, or conjugated to an antigen, which is bound to the antibody. The antibodies and fluorophores used should not interfere with the binding capacity of the antigen of interest and will depend on the sample and target antigen.
There are two main methods of immunofluorescence detection: Primary (or direct) and secondary (or indirect). Primary detection uses one primary antibody bound to a fluorophore, which then recognizes an epitope on the antigen of interest and can be visualized using a fluorescence microscope. Secondary detection uses two antibodies for detection. The first antibody is specific for the target antigen, and the second antibody is fluorophore conjugated and specific for the first antibody. The antigen-antibody-antibody-fluorophore complex can then be visualized using a fluorescence microscope.
RD253204A
Immunoprecipitation
Immunoprecipitation (IP) is a technique used to precipitate an antigen of interest out of solution via binding from an antibody conjugated to a solid substrate. There are different types of IP that target different types of proteins or other molecules. Individual protein IP targets one specific protein in a solution (usually containing other proteins). Protein complex immunoprecipitation (Co-IP) targets protein complexes instead of individual proteins. Chromatin immunoprecipitation (ChIP) is used in vivo to detect the location of DNA binding sites of a protein of interest. RNP immunoprecipitation (RIP) is used to detect ribonucleoproteins (RNPs) on RNA. There are two main methods of immunoprecipitation: Direct or Indirect. The direct method uses antibodies immobilized on a solid substrate that are specific for the target protein to collect the target protein. The indirect method uses antibodies that are specific for the target protein, but freely added to the sample. Following that, Protein A/G-linked beads are added, which binds both the antibody and target proteins. In both methods, the solid substrates (with proteins of interest) must then be isolated, typically either via centrifugation (with agarose beads) or a magnetic field (with superparamagnetic beads).
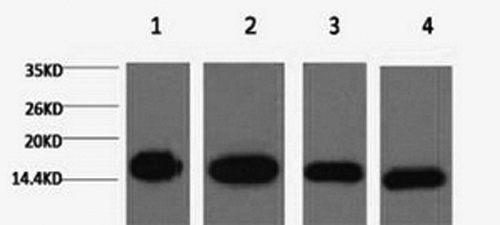
Western Blotting
The Western Blot (WB) is a technique used to detect and identify specific proteins in a sample. (Not to be confused with the Southern Blot for DNA detection and the Northern Blot for RNA detection.) Western Blotting has three main parts of the procedure: Separation of the specific protein of interest, transfer to a solid substrate, and labelling of the target protein with antibodies for visualization. In the first step, the proteins in the sample(s) are separated using gel electrophoresis, typically SDS polyacrylamide gel electrophoresis (SDS-PAGE). After separation, the proteins in the gel are then transferred onto a membrane (usually nitrocellulose or PVDF) in the same layout as they were on the gel. In the last step, labelling and visualization, typcailly an indirect method is used. The membrane is blocked to prevent any non-specific binding before being incubated with the primary antibody, which binds the protein of interest. After a wash step, a secondary antibody conjugated with a detection molecule specific for the primary antibody is added. Detection and visualization are achieved either by colorimetric, chemiluminescent, or radioactive methods.
ELISA
Enzyme-linked immunosorbent assay (ELISA) is a common assay that detects and quantifies the amount of analyte in a sample. There are four main styles of ELISA, but they all make use of antibody-antigen interactions for detection and quantitation. The sandwich format is the most popular because of its high specificity and sensitivity. The sandwich format involves ‘sandwiching’ the antigen of interest between two antibodies that are a matched pair. Other styles include Direct ELISA, Indirect ELISA, and Competitive Inhibition ELISA. In a direct ELISA, only one antibody is used to detect the antigen of interest that is bound to the well. The antibody is bound to a detection molecule. In an indirect ELISA, there is a two step detection method, where a primary antibody binds to the antigen of interest, and a secondary antibody conjugated to a detection molecule binds to the primary antibody. Competitive inhibition ELISAs provide inverse results by measuring the level of competition with an inhibitor antigen. Both the antigen of interest and inhibitor antigen are added at the same time, but only the inhibitor antigens can be bound by detection molecules, and therefore, detected. As a results, the higher the level of detection in competitive inhibition ELISAs, the lower the amount of target analyte.
To learn more about ELISA kit test principles, visit this blog post.
Flow Cytometry
Flow cytometry (FCM) is a technique used to detect and analyze groups of cells or other particles based on characteristics or attached labels. A flow cytometer instrument is necessary for this technique. The sample of cells or other particles is suspended in liquid medium and inserted into the instrument. The flow cytometer flows each cell or particle one at a time through a laser beam for detection and classification. When the cells or particles are sent though the beam, the light is scattered in such a way as to differentiate characteristics of the cells or particles. If the cells or particles are tagged with fluorescent markers, the light from the laser beam is also then emitted at different wavelengths. The data is collected and can then be further analyzed.

Intracellular Flow Cytometry
Intracellular Flow Cytometry (ICFCM) is a similar technique to Flow Cytometry but used to analyze molecules inside the cell such as phosphorylated signaling proteins or cytokines. In order to tag and visualize proteins inside the cell, the cells in the sample should be treated with a protein transport inhibitor, then fixed in suspension and permeabilized before a detection antibody with fluorophore can be added. Following this, the sample can then be analyzed with a flow cytometer.
To browse our full catalogue of antibodies, visit our Antibodies Product Page.
For more information, visit our FAQ or Troubleshooting pages, or contact us for assistance.